Table of Contents >> Show >> Hide
- What is Angelman syndrome?
- Core signs and symptoms of Angelman syndrome
- What causes Angelman syndrome?
- How Angelman syndrome is diagnosed
- Treatments for Angelman syndrome
- Long-term outlook and quality of life
- Are new treatments on the horizon?
- Conclusion
- Common family and caregiver experiences with Angelman syndrome
- SEO Tags
Angelman syndrome (AS) is one of those conditions that sounds obscure until it lands in a real family’s life and suddenly becomes the center of every question, appointment, and late-night search. It is rare, lifelong, and deeply tied to brain development. But it is also a condition that clinicians understand far better today than they did a generation ago. Families now have clearer diagnostic pathways, stronger therapy plans, better seizure management, more communication tools, and growing hope from targeted research.
This guide explains what Angelman syndrome is, what causes it, how it is diagnosed, and which treatments can make a meaningful difference. It also looks at what daily life can actually feel like, because medical definitions matter, but so do practical realities like sleep, school, feeding, communication, and the emotional whiplash of trying to help a child thrive.
What is Angelman syndrome?
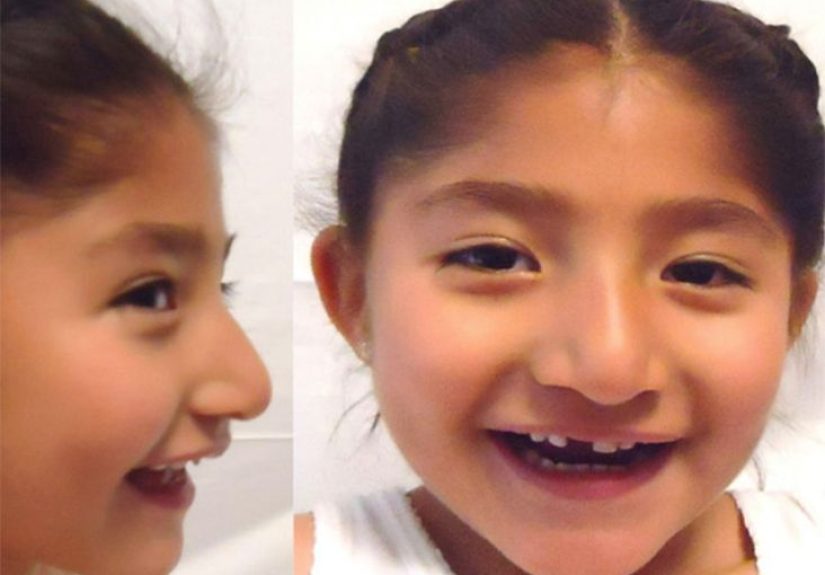
Angelman syndrome is a rare neurogenetic disorder that primarily affects the nervous system. In plain English, that means it is caused by a genetic problem that changes how the brain develops and functions. Children and adults with AS usually have significant developmental delay, intellectual disability, very limited speech or no functional spoken language, movement and balance problems, and a recognizable behavioral profile that may include frequent smiling, laughter, excitability, and hand-flapping.
AS is not usually obvious at birth. Many babies look healthy in the newborn period, which can make the early months confusing for parents. Developmental delays often become noticeable between 6 and 12 months. A baby may not babble much, may not crawl on schedule, or may seem to have trouble with feeding, motor coordination, or sleep. As the child gets older, the pattern becomes clearer: speech is severely affected, walking may be stiff or shaky, seizures may develop, and communication often relies more on gestures, facial expression, devices, and learned routines than on spoken words.
Although Angelman syndrome is serious, it is not a progressive neurodegenerative disease in the usual sense. People with AS do not typically lose skills in the way seen in some other neurologic conditions. Many can make steady gains over time, especially with early intervention, structured therapy, and strong support at home and school. That is an important point, because the diagnosis is life-changing, but it is not the end of growth, learning, joy, or personality. Far from it.
Core signs and symptoms of Angelman syndrome
Developmental and neurologic features
The hallmark symptoms of Angelman syndrome include delayed development, severe speech impairment, intellectual disability, and problems with movement and balance. Many children have ataxia, which means unsteady or poorly coordinated movement. Walking may look stiff-legged, jerky, or wide-based. Tremulous limb movements can also appear. Seizures are common, and some children have abnormal EEG patterns even before the full clinical picture becomes obvious.
Behavior and communication
One of the most recognizable features of AS is an unusually happy, excitable demeanor. Many children smile often, laugh easily, and show strong social interest. That cheerful presentation can be charming, but it should not fool anyone into thinking the condition is mild. Underneath the smiles can be major communication barriers, sensory challenges, frustration, sleep disruption, and intense caregiving needs.
Speech is usually the area with the greatest impairment. Some individuals may learn a few words, but many remain minimally verbal. That does not mean they have nothing to say. In fact, many people with Angelman syndrome understand far more than they can express. This is why speech therapy for AS often focuses less on perfect pronunciation and more on functional communication, especially through augmentative and alternative communication, or AAC.
Other common issues
AS can also involve feeding difficulties in infancy, reflux, constipation, sleep disturbance, strabismus, scoliosis, and a smaller-than-expected head size by early childhood. Sleep can be especially difficult for families. Some children wake repeatedly, rise very early, or seem to treat bedtime as a polite suggestion rather than a rule. That may sound funny on paper, but at 3:14 a.m. it is less “quirky child behavior” and more “everyone in this house needs coffee and a care plan.”
What causes Angelman syndrome?
The biology of Angelman syndrome centers on a gene called UBE3A, located on chromosome 15. Most people inherit one copy of this gene from each parent. In many tissues, both copies can work. But in certain brain cells, the maternal copy is the one that matters most. When that maternal UBE3A function is missing or disrupted, Angelman syndrome can result.
This is why AS is often described as a disorder of genomic imprinting. That phrase sounds like something a genetics professor mutters while drawing confusing arrows on a whiteboard, but the practical idea is simpler: in the brain, the body usually depends on the mother’s working copy of UBE3A. If that copy is deleted, altered, or silenced, the nervous system does not get the instructions it needs.
The main genetic mechanisms
There is no single cause behind every case of Angelman syndrome. Instead, several genetic mechanisms can lead to the same broad diagnosis:
1. Maternal deletion of chromosome 15q11.2-q13. This is the most common cause and accounts for about 70% of cases. In these children, a piece of the mother’s chromosome 15 that includes UBE3A is missing.
2. A pathogenic variant in the maternal UBE3A gene. In roughly 10% to 20% of cases, the gene is present but changed in a way that disrupts its function.
3. Paternal uniparental disomy (UPD). In this situation, the child inherits both copies of the relevant chromosome region from the father and none from the mother.
4. Imprinting defects. Sometimes the maternal chromosome is present, but the “parent-of-origin” marking does not work correctly, so the brain does not use the maternal UBE3A copy as it should.
5. Unknown causes. In a smaller group of people who clearly look clinically consistent with AS, currently available testing still does not identify the exact mechanism.
Most cases are not caused by anything the parents did or did not do. This is not a parenting issue, not a pregnancy “mistake,” and not something caused by a missed vitamin, a stressful week, or a badly timed slice of pizza. It is a genetic event. That distinction matters, especially for families carrying unnecessary guilt.
How Angelman syndrome is diagnosed
Diagnosis usually begins with clinical suspicion. A doctor may start considering AS when a child has developmental delay, little or no speech, balance problems, a happy or excitable demeanor, seizures, or microcephaly. The challenge is that these signs overlap with other neurodevelopmental conditions, which is why Angelman syndrome can be missed early on.
Genetic testing matters
The diagnostic workup typically includes molecular genetic testing rather than relying on symptoms alone. DNA methylation analysis is often the first test because it can identify many cases caused by maternal deletion, paternal UPD, or imprinting defects. If methylation testing is normal but suspicion remains high, clinicians may move on to additional studies such as UBE3A sequencing and chromosomal microarray.
Getting the exact genetic mechanism is important for more than naming the condition. It can help predict recurrence risk, guide genetic counseling for the family, and sometimes explain why one child’s symptoms are milder or more severe than another’s. It also helps families enter research studies more accurately, since some clinical trials and future therapies may target specific subtypes.
Treatments for Angelman syndrome
There is currently no cure that reverses Angelman syndrome. Treatment focuses on managing symptoms, improving function, supporting development, and protecting quality of life. In other words, care is supportive, multidisciplinary, and highly individualized.
Seizure treatment
Seizures are common in AS, so neurologic care is a major part of treatment. Anti-seizure medications are often used, although seizure control can be tricky because more than one seizure type may occur. Some individuals benefit from dietary approaches such as ketogenic or low-glycemic strategies when seizures are difficult to manage. Families may also need rescue medications and a clear action plan for prolonged seizures.
Speech therapy and AAC
Speech therapy is essential, but not in the narrow “say this word clearly” sense. For many people with AS, the real goal is communication, not conventional speech. AAC tools such as picture cards, communication boards, sign systems, and speech-generating devices can be life-changing. Starting these supports early can reduce frustration, improve participation, and help the child show what they know.
Physical and occupational therapy
Physical therapy can help with gait, balance, coordination, strength, posture, and mobility. Occupational therapy supports fine-motor skills, self-care tasks, sensory regulation, and adaptive strategies for everyday activities. These therapies do not “fix” Angelman syndrome, but they can absolutely improve independence, safety, and confidence.
Sleep and behavior support
Sleep problems are incredibly common. Families may need a combination of behavior strategies, good sleep hygiene, environmental safety measures, and sometimes medication such as melatonin under medical guidance. Behavioral support can also help with hyperactivity, impulsivity, self-injury, and routines that affect school and family life.
Feeding, GI, vision, and orthopedic care
Treatment may also involve reflux management, constipation care, feeding therapy, special nipples or positioning strategies in infancy, ophthalmology care for strabismus, and monitoring for scoliosis or ankle and foot problems. In some cases, braces, adaptive chairs, or surgery may be considered. This is why families often end up with a care team that includes neurology, genetics, developmental pediatrics, gastroenterology, orthopedics, ophthalmology, rehab specialists, therapists, and school professionals. It is a lot. Helpful, necessary, and often exhausting, but still a lot.
Long-term outlook and quality of life
People with Angelman syndrome often live close to a typical lifespan, but they usually need lifelong support. Independence varies widely. Some individuals can learn many self-help skills and enjoy strong social engagement, while others need intensive supervision throughout life. Early diagnosis, consistent therapy, careful seizure management, and good communication support can make a major difference in day-to-day function.
Importantly, the outlook is not defined only by medical limitations. Many families describe their loved one with AS as affectionate, expressive, socially connected, and full of personality. The challenges are real, but so is the capacity for connection, learning, humor, and joy.
Are new treatments on the horizon?
Yes, but with an important asterisk. Research is moving fast, especially in therapies aimed at restoring UBE3A activity in the brain. Investigational approaches include antisense oligonucleotides, gene therapy, and other strategies designed to unsilence or replace the missing maternal function. These developments are exciting and scientifically meaningful.
Still, families should hear the full sentence, not just the hopeful half. These approaches remain experimental. They are not yet routine standard care, and they do not replace the therapies people with AS need right now. Today’s best treatment plan is still a strong combination of medical care, seizure management, developmental therapy, communication support, and practical family resources.
Conclusion
Angelman syndrome is a rare but recognizable neurogenetic disorder linked to loss of maternal UBE3A function in the brain. It affects communication, movement, cognition, sleep, and behavior, and it often becomes visible in infancy as developmental milestones begin to drift off schedule. Diagnosis depends on clinical awareness plus genetic testing, and treatment is centered on supportive care rather than cure.
That may sound heavy, because it is. But it is not hopeless. The combination of early diagnosis, coordinated therapy, AAC, seizure care, and strong family support can dramatically improve everyday life. And as research advances, the story of Angelman syndrome is shifting from “rare and misunderstood” to “rare, better defined, and actively targeted.” For families, that is more than a scientific update. It is a reason to keep going.
Common family and caregiver experiences with Angelman syndrome
Families often describe the first phase of Angelman syndrome as a season of uncertainty. A baby may be sweet, alert, and seemingly fine, but something starts to feel off. Milestones are late. Babbling does not really happen. Sitting, crawling, or feeding may seem harder than expected. Parents bring up concerns and are sometimes told to “wait and see,” which can be frustrating because their instincts are already waving a bright little red flag. The eventual diagnosis can be painful, but it can also bring relief. At least the uncertainty finally has a name.
Another common experience is the long learning curve around communication. Many caregivers say one of the hardest parts is realizing their child understands more than other people assume, while still having very limited speech. This gap can lead to underestimation by teachers, relatives, or even strangers. Families often become fierce advocates for AAC, visual supports, and communication devices. The first time a child clearly requests a favorite snack, chooses an activity, or signals “all done” through a board or device, it can feel like a door quietly swings open in the middle of the house.
Sleep is another major theme in family life with AS. Caregivers frequently talk about interrupted nights, unusual sleep-wake cycles, and the exhaustion that comes from always being half on call. It is not just a child waking up early. It can be a safety issue, especially if the child is mobile, impulsive, or fascinated by water, climbing, or exploration. Families often end up redesigning bedrooms, changing evening routines, and becoming accidental experts in sleep hygiene, white noise, blackout curtains, medication timing, and how to function on less rest than any adult should reasonably be expected to survive on.
Seizures can add another layer of stress. Even when they are eventually controlled, many families carry ongoing anxiety around illness, medication adjustments, and emergency plans. Some describe always knowing where the rescue medicine is, always tracking symptoms, and always calculating whether a strange movement is “just movement” or something more serious. That constant vigilance is tiring, but it also reflects how skilled families become. Over time, many caregivers develop an impressive working knowledge of neurology, therapy systems, insurance rules, educational law, and adaptive equipment. They did not ask for this major, but they definitely earned the degree.
At the same time, families often describe deep joy in the personality of their loved one. They talk about expressive faces, contagious laughter, huge social warmth, and strong emotional presence. Progress may not always look typical, but it matters just as much. A better night of sleep, a safer gait, a successful school routine, using a device to say “more,” or learning to participate in dressing and mealtime can be huge wins. These moments may seem small to outsiders, yet inside a family they can feel monumental.
As children with AS grow older, many families begin thinking beyond childhood services and into adulthood, long-term care, and quality of life across decades. That shift can be emotional. Parents are not just managing a diagnosis; they are planning a future. They want safety, dignity, communication, meaningful routines, and community. The experience of living with Angelman syndrome is rarely simple, but it is not defined by difficulty alone. It is also shaped by resilience, adaptation, advocacy, and the steady belief that connection and progress are always worth pursuing.





