
Table of Contents >> Show >> Hide
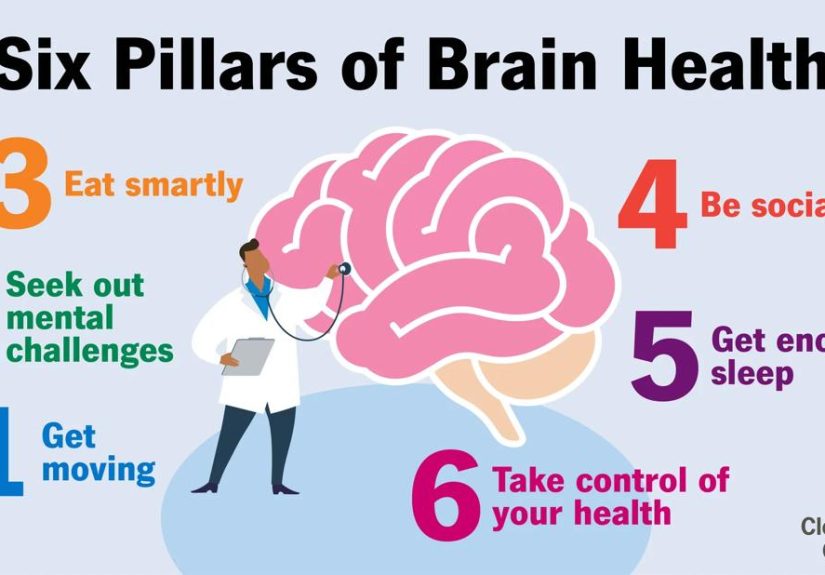
- What inherited hemophilia is (and what it isn’t)
- How hemophilia is inherited: the X-chromosome story
- Testing and diagnosis: how inherited hemophilia is confirmed
- Treatment: the modern toolbox (and how doctors choose the right tool)
- Gene therapy: “one-time” treatment, real-world caveats
- Hemophilia treatment centers: why specialized care matters
- Outlook: what life can look like with inherited hemophilia
- FAQ
- Experiences from the hemophilia community (real-life patterns you’ll hear again and again)
- SEO Tags
If blood clotting were a band, hemophilia is what happens when the drummer doesn’t show up. The rest of the group (platelets, blood vessels, and other clotting proteins) might try their best, but without enough factor VIII (hemophilia A) or factor IX (hemophilia B), clots can form too slowly or fall apart too easily.
The good news: inherited hemophilia is one of the best examples of how modern medicine can turn a serious genetic condition into something people manageoften very successfullyover a lifetime. Between better testing, safer factor products, new “non-factor” medications, and even one-time gene therapy options for some adults, the outlook today is dramatically different than it was even a couple decades ago.
In this guide, we’ll walk through how inherited hemophilia is passed down, how testing works (for babies, kids, and carriers), what treatment looks like in 2025, and what day-to-day life and long-term health can look likewith fewer surprises and more confidence.
What inherited hemophilia is (and what it isn’t)
Inherited hemophilia is a genetic bleeding disorder caused by changes (variants) in the genes that help the body make clotting factor VIII or IX. When factor levels are low, bleeding can last longer than expected, especially after injuries, dental work, or surgery. In more severe cases, bleeding can happen “out of nowhere,” including into joints and muscles.
It’s worth saying out loud: inherited hemophilia is not caused by anything someone did wrong. It isn’t contagious, it isn’t a “weak blood” problem, and it isn’t a character flaw. It’s biologyspecifically, genetics.
Hemophilia A vs. Hemophilia B
- Hemophilia A: factor VIII (8) deficiency
- Hemophilia B: factor IX (9) deficiency
Doctors also describe hemophilia by severity, based on factor activity levels:
- Severe: very low factor activity (often <1%) and higher risk of spontaneous bleeding
- Moderate: bleeding typically with injuries, surgery, or significant trauma
- Mild: bleeding may show up mainly with procedures, major injuries, or certain life events (like childbirth or major dental work)
How hemophilia is inherited: the X-chromosome story
Most inherited hemophilia A and B follow an X-linked recessive inheritance pattern. Here’s the short version:
- Males typically have one X and one Y chromosome (XY). If their single X has the hemophilia variant, they can have hemophilia.
- Females typically have two X chromosomes (XX). If one X has the variant, they’re often called carriers, but some carriers also have bleeding symptoms (more on that soon).
Common family scenarios
Scenario 1: Mom is a carrier, Dad does not have hemophilia.
- Each son has a 50% chance of having hemophilia.
- Each daughter has a 50% chance of being a carrier.
Scenario 2: Dad has hemophilia, Mom is not a carrier.
- Sons do not inherit dad’s X chromosome, so they won’t inherit hemophilia from him.
- All daughters inherit dad’s X chromosome, so they will be carriers (and some may have symptoms).
Scenario 3: No family history… and then hemophilia appears.
This happens more often than many people expect. A new genetic change can occur for the first time in a child (a “new mutation”), meaning there may be no known hemophilia in prior generations.
Carriers can have symptomsyes, really
Some people hear “carrier” and assume it means “no symptoms.” But many carriers have factor levels lower than average due to natural differences in X-chromosome activity (including skewed X-inactivation). That can lead to easy bruising, heavy menstrual bleeding, postpartum bleeding risk, or extra bleeding after procedures. If you’re a known or possible carrier, testing isn’t just about family planningit can be about your health, too.
Testing and diagnosis: how inherited hemophilia is confirmed
Hemophilia testing usually has two goals:
(1) confirm a diagnosis (type and severity) and
(2) understand the genetic variant in the family (for carrier testing, future pregnancies, and personalized care).
Step 1: Screening blood tests
If bleeding history raises suspicion, doctors often start with screening labs that check how well the blood is clotting. A common clue for hemophilia is a prolonged aPTT (activated partial thromboplastin time), though mild cases can sometimes have near-normal screening results.
Step 2: Factor assays (the main event)
The most important test is a clotting factor assay, which measures how much factor VIII and factor IX activity is present. This confirms whether it’s hemophilia A or B and helps define severity (mild/moderate/severe).
Step 3: Genetic testing (the “why” behind the numbers)
Genetic testing looks for variants in the F8 gene (hemophilia A) or F9 gene (hemophilia B). It can be used to:
- Confirm the inherited type in a family
- Identify carriers (including women and girls who may have symptoms)
- Support prenatal or newborn testing decisions
- Help anticipate inhibitor risk in some situations (especially based on certain variant types)
Testing for inhibitors (when treatment stops working like it should)
Some people develop antibodies called inhibitors that neutralize factor VIII or factor IX, making standard factor infusions less effective. Inhibitor testing is a key part of follow-up careespecially if bleeding episodes increase, factor doses seem to “wear off” too quickly, or someone is newly starting factor therapy.
Carrier testing and family planning options
If hemophilia runs in your family, genetic counseling can help map out testing choices. Options may include:
- Carrier testing for relatives who may carry the family variant
- Prenatal testing (such as CVS or amniocentesis) in some pregnancies
- Newborn testing soon after birth (especially when there’s known family history)
- Preimplantation genetic testing (PGT) with IVF for families who choose that route
These decisions are personal. The “right” choice is the one that fits the family’s values, health situation, and access to specialized care.
Treatment: the modern toolbox (and how doctors choose the right tool)
Hemophilia treatment usually falls into two big strategies:
- On-demand treatment: treat bleeds as they happen
- Prophylaxis: prevent bleeds before they happen (now a common standard for severe hemophilia)
1) Factor replacement therapy (classic, effective, still widely used)
Factor replacement means infusing factor VIII or IX into a vein to restore clotting ability. Products may be:
- Recombinant (made in a lab)
- Plasma-derived (from donated human plasma, processed and treated for safety)
- Extended half-life versions designed to last longer, reducing infusion frequency for some people
Factor replacement can be used both on-demand and as prophylaxis. Many families also learn home infusion skillsbecause waiting rooms are not a hobby anyone asked for.
2) Non-factor therapy (helping the body clot without replacing factor)
The last several years have brought a wave of medications that work differently than factor replacement. These options can reduce bleeds while avoiding frequent IV infusionsoften a big quality-of-life upgrade.
Emicizumab (Hemlibra)
Emicizumab is a “factor VIII mimetic” antibody given under the skin that helps bridge part of the clotting process. It’s used as prophylaxis for hemophilia A and can be an option even when inhibitors are present (depending on the care plan).
“Rebalancing” agents (newer options)
These therapies tilt the clotting system toward better balance by reducing natural anticoagulant pathways or blocking certain proteins. In the U.S., several have FDA approvals for routine prophylaxis in specific groups, typically ages 12 and older, with details depending on inhibitors and the medication:
- Fitusiran (Qfitlia): an antithrombin-lowering therapy given by subcutaneous injection, approved for hemophilia A or B (with or without inhibitors) in patients 12+.
- Concizumab (Alhemo): an anti-TFPI therapy given by subcutaneous injection, initially approved for patients 12+ with inhibitors; labeling has expanded as evidence has grown.
- Marstacimab (Hympavzi): another anti-TFPI approach given weekly by subcutaneous injection, approved for hemophilia A or B without inhibitors in patients 12+.
These medicines are excitingbut they also come with specific monitoring needs and safety considerations. That’s why they’re usually managed by clinicians experienced in bleeding disorders, often at hemophilia treatment centers.
3) Treatment for mild hemophilia: sometimes less is more
People with mild hemophilia may not need regular prophylaxis. Instead, treatment may focus on procedures and higher-risk moments. Common options include:
- Desmopressin (DDAVP) for some people with mild hemophilia A (it helps the body release stored factor VIII). It’s not used for hemophilia B.
- Antifibrinolytics (like tranexamic acid or aminocaproic acid), which help keep clots from breaking downespecially useful for dental work, nosebleeds, and mouth bleeding.
- Topical and local measures (for example, dental approaches, nasal packing strategies, and careful procedure planning)
Think of mild hemophilia management like carrying an umbrella. You don’t need it every day, but when the forecast changes, you’re very glad it’s in your bag.
4) Inhibitors: the plot twist that changes the plan
Inhibitors can make standard factor replacement less effective. When inhibitors develop, treatment plans may include:
- Bypassing agents to help clotting occur through alternate pathways
- Non-factor prophylaxis options, depending on hemophilia type and inhibitor status
- Immune tolerance induction (ITI) in some cases (a strategy that aims to train the immune system to stop reacting to factor)
- Closer monitoring and customized emergency plans
If hemophilia is a chess game, inhibitors are like someone swapping your queen for a potato mid-match. You can still winbut you’ll need a different strategy and a specialist who has seen this before.
Gene therapy: “one-time” treatment, real-world caveats
Gene therapy for hemophilia aims to deliver a working copy of the clotting factor gene to liver cells, allowing the body to produce more factor on its own. In the U.S., FDA-approved gene therapies include:
Hemophilia B gene therapy: Hemgenix
Hemgenix is a one-time IV infusion approved for adults with hemophilia B who use factor IX prophylaxis or have serious bleeding history (such as life-threatening bleeding or repeated serious spontaneous bleeding). It is designed to raise factor IX levels for an extended period.
Hemophilia A gene therapy: Roctavian
Roctavian is a one-time IV infusion approved for adults with severe hemophilia A who meet specific eligibility criteria (including no pre-existing antibodies to the AAV5 vector, measured by an FDA-approved test). It is intended to increase factor VIII activity and reduce bleeding and the need for routine factor infusions in some patients.
What gene therapy doesn’t do
- It is not currently a routine option for children.
- It doesn’t “edit” DNA in a way that guarantees permanent correction in every cell.
- Results can vary from person to person, and factor levels may change over time.
- Follow-up monitoring (including liver-related labs) is part of the package.
Gene therapy is best viewed as a major optionnot magic. For the right person, it can be life-changing. For others, current therapies may still be the better fit.
Hemophilia treatment centers: why specialized care matters
Hemophilia is more than “just bleeding.” It can affect joints, muscles, school sports decisions, dental planning, mental stress, and how families prepare for emergencies. That’s why many people benefit from care at a Hemophilia Treatment Center (HTC), where teams often include hematology specialists, nurses, physical therapists, social workers, and care coordinators.
HTCs can be especially helpful for:
- Choosing and adjusting prophylaxis plans
- Inhibitor monitoring and management
- Planning surgery and dental procedures
- Helping carriers who have symptoms get properly evaluated
- Teaching home infusion skills and building emergency action plans
Outlook: what life can look like with inherited hemophilia
With consistent treatment and good follow-up, many people with inherited hemophilia can expect to:
- Attend school, work, travel, and play sports (with smart precautions)
- Reduce joint damage through early prophylaxis and physical therapy
- Handle surgeries and dental work safely with planning
- Build long-term routines that make hemophilia one part of lifenot the headline
Protecting joints and staying active
Joint bleeds are a major concern, especially in severe hemophilia. Preventing bleeds early helps protect joints over time. Many clinicians encourage regular activity because strong muscles support jointsjust with a thoughtful approach to contact risk. Swimming, biking with protective gear, strength training with guidance, and many team sports can be possible depending on severity and prophylaxis.
School, sports, and the “please don’t panic” plan
Families often create a simple school plan: who to call, where factor (or rescue meds) are stored if appropriate, what symptoms should trigger urgent action, and how to handle injuries. A medical ID bracelet or phone medical ID can be a small thing that makes a big difference when time matters.
Carriers, menstruation, and pregnancy
If you’re a carrieror could befactor level testing and bleeding history matter. Heavy menstrual bleeding and postpartum bleeding risk deserve real attention and real solutions (not “that’s just how periods are”). Many carriers benefit from individualized planning for childbirth and procedures, ideally with coordination between obstetrics and hematology.
FAQ
Can someone have hemophilia with no family history?
Yes. New genetic changes can occur, and hemophilia may be diagnosed in a child even when no relatives were previously known to have it.
Is genetic testing always necessary?
Not always to manage day-to-day bleeding risk (factor levels often guide that), but genetic testing can be extremely helpful for carrier testing, future pregnancies, confirming the family variant, and long-term planning.
Does prophylaxis mean “no bleeds ever”?
Prophylaxis can dramatically reduce bleeding episodes, especially spontaneous bleeds, but it doesn’t make someone invincible. Injuries, procedures, and individual response still matter, and treatment plans often include “breakthrough bleed” instructions.
What’s the biggest predictor of a good long-term outcome?
Consistent access to appropriate therapy, early prevention of joint bleeds, and a care team that knows hemophilia well. In other words: the right plan, started early, and adjusted as life changes.
Medical note: This article is for general education and is not a substitute for professional medical advice. If you suspect hemophilia or manage a confirmed diagnosis, a hematology teamideally through an HTCcan tailor testing and treatment to the individual.
Experiences from the hemophilia community (real-life patterns you’ll hear again and again)
You can read all the lab values in the world and still feel unprepared the first time hemophilia becomes part of your daily routine. What many families and patients describe is a journey that starts with confusion (“Why won’t the bleeding stop like it does for everyone else?”) and gradually becomes a set of skillspractical, emotional, and surprisingly empowering.
One of the most common “early chapters” is the diagnostic whirlwind. Parents often talk about the first major bruise that looks too dramatic for the event, a prolonged bleed after a routine procedure, or a baby who seems to swell at a joint after minor bumps. For some families, the diagnosis comes quickly because hemophilia is already known in the family. For others, there’s a strange mix of relief and fear: relief because there’s finally an explanation, and fear because the word “genetic” sounds permanent (even though treatment has come a long way).
Then comes the “skills era.” If factor replacement is part of the plan, people often describe learning home infusion like learning to drive: intimidating at first, then gradually becoming normal. There’s the first time holding supplies with shaky hands, the first successful infusion that feels like winning a tiny trophy, and the moment you realize you’re not waiting helplessly for helpyou’re part of the care team. Many families also describe the value of an HTC nurse who teaches without judgment, answers “one more question” for the fifteenth time, and treats confidence as a medical outcome (because it is).
For teens, experiences often shift toward independence and identity. Some describe the awkward phase of explaining hemophilia to friends without sounding like a medical textbook. Others describe the “quiet pressure” of wanting to do everything peers dosports, travel, sleepoverswhile also knowing they need a plan. The best stories here aren’t about saying “no” to life; they’re about saying “yes, and here’s how.” Yes to basketball, and you wear the right protective gear, keep prophylaxis consistent, and know what to do if you take a hard hit. Yes to a school trip, and you coordinate meds and carry medical info. Hemophilia becomes less of a cage and more of a checklist.
Many adults with hemophilia describe a different turning point: moving from reacting to bleeding to preventing it. People who grew up before modern prophylaxis sometimes talk about joint pain and limitations that shaped their routines. In contrast, younger patients on early prophylaxis often describe a childhood with far fewer joint bleedsand a future that looks a lot more open. That contrast can bring complicated feelings in families: gratitude for modern therapy, frustration about access barriers, and a strong sense of community advocacy (“If this works, everyone should be able to get it”).
Carriers often share a specific kind of experience: not being taken seriously at first. Some describe years of heavy periods or easy bruising being dismissed, only to later discover that their factor levels are lower than expected. The most validating moments often come when a hematology-informed clinician connects the dots and offers real optionstreatment plans for procedures, strategies for menstrual bleeding, and coordinated pregnancy planning when relevant.
Across ages, one theme shows up constantly: hemophilia is easier to manage when the plan is simple, written down, and shared. Families often keep a one-page emergency guide, store medication info in phones, and build relationships with providers who know them. And yespeople still crack jokes. Humor shows up not because hemophilia is funny, but because laughter is a practical coping tool. If you can laugh while organizing infusion supplies at 6 a.m., you can handle a lot.