
Table of Contents >> Show >> Hide
Langerhans cell histiocytosis, usually shortened to LCH, is one of those conditions that sounds like it was named during a game of medical Scrabble. Unfortunately, the disease itself is very real. LCH is a rare disorder in which abnormal immune cells build up in tissues and trigger inflammation, damage, and sometimes scarring. It can affect one body part, such as a bone, or show up in several organs at once and act far more aggressively.
What makes LCH tricky is that it does not come with one tidy, obvious symptom list. One person may have a painful bone lesion. Another may develop a stubborn rash. Someone else may show up with breathing problems, excessive thirst, or repeated ear infections that will not quit. In other words, LCH is a master of disguise. It can look like eczema, infection, arthritis, or even cancer before doctors finally connect the dots.
This guide breaks down the symptoms of Langerhans cell histiocytosis, what researchers know about its causes, how doctors diagnose it, and the most common LCH treatment options used today. The goal is simple: clear, useful information without the fog machine of medical jargon.
What Is Langerhans Cell Histiocytosis?
LCH is a rare disease involving abnormal growth and accumulation of cells that resemble Langerhans cells, which are part of the body’s immune system. Under normal circumstances, these cells help the immune system recognize threats. In LCH, similar cells multiply in the wrong way, collect in tissues, and create lesions or inflammation.
Experts now generally view LCH as a clonal myeloid neoplasm, meaning it behaves more like a disorder driven by abnormal cell growth than a simple overreaction of the immune system. At the same time, inflammation plays a huge role in how the disease behaves. That is why LCH sits in an awkward middle seat between “immune condition” and “cancer-like disorder.” It is rare, complicated, and not particularly interested in fitting neatly into one box.
LCH can affect both children and adults, although it is more common in children, especially in early childhood. In kids, the disease often involves bone, skin, lymph nodes, or the pituitary gland. In adults, the lungs may be involved more often, especially in people who smoke.
Symptoms of LCH
LCH symptoms vary depending on which organs are affected and whether the disease is limited to one site or spread across several systems. That is why one person’s version of LCH can look nothing like another’s.
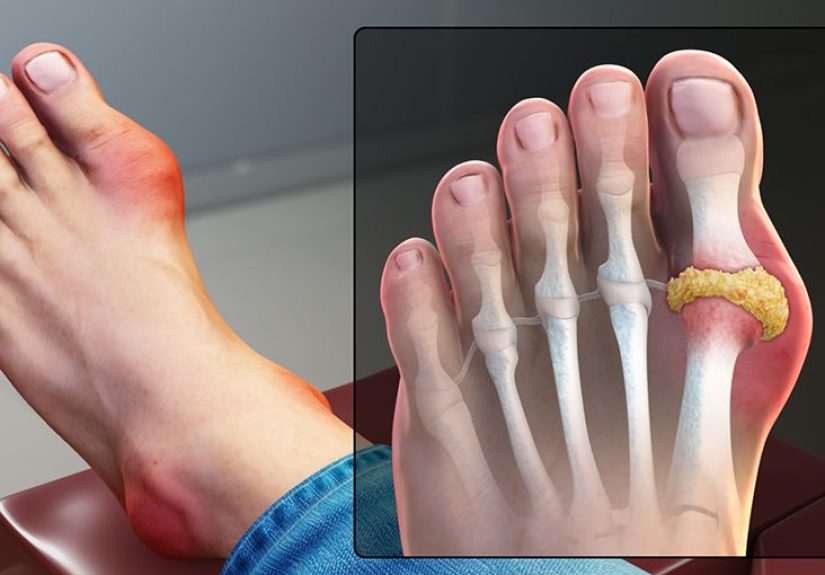
Bone Symptoms
Bone is one of the most common places for LCH to appear, especially in children. Lesions may form in the skull, ribs, jaw, spine, arms, or legs. Common symptoms include:
- Localized bone pain or tenderness
- Swelling over the affected area
- A limp or reduced movement
- Loose teeth or jaw pain if facial bones are involved
- Fractures in weakened bone
A child with LCH in the skull may develop a tender bump that seems to appear out of nowhere. An adult with spinal involvement may notice back pain that does not improve with rest. This is where LCH can start acting like a bad impersonator of sports injury, dental trouble, or growing pains.
Skin Symptoms
LCH can affect the skin and scalp, sometimes making it look like severe cradle cap, eczema, or a persistent rash. Symptoms may include:
- Scaly, greasy, or crusted rash on the scalp
- Red or brown bumps on the trunk or diaper area
- Ulcerated skin lesions
- Persistent rash that does not respond to usual treatment
Because the rash can mimic common skin conditions, diagnosis may be delayed. What looks like stubborn dermatitis may turn out to be a clue pointing to something deeper.
Pituitary and Hormonal Symptoms
When LCH affects the pituitary gland or nearby brain structures, it can interfere with hormone control. One of the best-known complications is diabetes insipidus, which is not the same thing as diabetes mellitus. Symptoms may include:
- Extreme thirst
- Frequent urination
- Bedwetting in a previously dry child
- Growth delay
- Puberty changes or other hormone-related issues
These symptoms can be easy to miss at first. Parents may think a child is just drinking more water than usual. Adults may blame stress, diet, or a very committed water bottle habit.
Lung Symptoms
Pulmonary LCH occurs more often in adults and is strongly linked to smoking. Symptoms may include:
- Dry cough
- Shortness of breath
- Chest pain
- Fatigue
- Collapsed lung in some cases
Some adults have lung disease discovered on imaging before symptoms become obvious. Others do not learn about it until a sudden pneumothorax, or collapsed lung, sends them to the emergency room.
Systemic or Multisystem Symptoms
When LCH affects multiple organs, symptoms may be broader and more serious. These can include:
- Fever
- Weight loss
- Swollen lymph nodes
- Enlarged liver or spleen
- Abdominal swelling
- Jaundice
- Recurrent ear drainage or infections
- Fatigue and irritability
Doctors often pay close attention when so-called “risk organs,” such as the liver, spleen, or bone marrow, are involved, because these cases may require more intensive treatment.
What Causes LCH?
The short version is this: the exact cause of Langerhans cell histiocytosis is not fully understood, but researchers know much more now than they did in the past. LCH is associated with mutations that activate the MAPK signaling pathway, which helps control cell growth and survival. One of the most well-known is the BRAF V600E mutation. Other cases may involve changes in genes such as MAP2K1.
These mutations do not usually mean a person “inherited” LCH from a parent in the classic sense. In many cases, the mutations appear during cell development rather than being passed down through families. That means LCH is generally not considered a contagious disease, and it is not caused by anything a parent did or did not do.
There are, however, some associations worth knowing:
- Smoking is strongly linked with pulmonary LCH in adults.
- Immune dysregulation and inflammation appear to contribute to how the disease behaves.
- Genetic mutations drive abnormal cell growth in many cases.
So, if you are looking for one neat cause with a bow on top, medicine unfortunately does not have that yet. The best current explanation is that LCH develops from abnormal immune precursor cells with mutations that push them into unhealthy growth and tissue infiltration.
How LCH Is Diagnosed
Because LCH can imitate so many other conditions, diagnosis usually requires a mix of clinical suspicion, imaging, and tissue testing. Doctors may begin with a physical exam and a close review of symptoms, then move on to targeted studies such as:
- X-rays for bone lesions
- CT scans or MRI for detailed imaging
- PET scans in some cases to look for active disease sites
- Blood tests to check organ function and inflammation
- Endocrine testing if hormone issues are suspected
- Lung function tests for pulmonary involvement
The most important step is often a biopsy. A tissue sample allows pathologists to identify the characteristic abnormal cells and confirm the diagnosis using special markers such as CD1a and langerin (CD207). Without biopsy confirmation, LCH can be mistaken for several other illnesses.
Once LCH is confirmed, doctors usually perform a staging-style workup to see whether the disease is limited to one organ or affecting multiple systems. That matters a great deal because treatment planning depends heavily on disease extent.
Treatment for Langerhans Cell Histiocytosis
LCH treatment depends on where the disease is located, how extensive it is, how old the patient is, and whether important organs are involved. Some cases are relatively mild and localized. Others need systemic therapy and long-term monitoring.
Treatment for Single-System LCH
If LCH affects only one area, especially one bone lesion, treatment may be less intense. Options can include:
- Surgical curettage or removal of the lesion
- Steroid injection into a bone lesion
- Topical therapies for limited skin disease
- Observation in carefully selected cases
- Radiation therapy in rare situations when lesions are hard to manage otherwise
Single-site disease often has an excellent outlook, although recurrence can still happen. Even when treatment seems straightforward, follow-up matters.
Treatment for Multisystem LCH
When multiple organs are involved, treatment often includes systemic therapy. In children, common first-line regimens have included combinations such as vinblastine and prednisone. Depending on the situation, doctors may also use medicines such as:
- Cytarabine
- Cladribine
- Clofarabine in selected refractory cases
- Other chemotherapy or immunomodulating approaches in specialized centers
For patients with relevant mutations, especially in relapsed or difficult-to-treat disease, targeted therapy may be an option. Drugs that inhibit BRAF or MEK have changed the conversation for some patients with mutation-driven disease. These treatments can be highly effective, but they require careful specialist oversight because they are not casual over-the-counter vitamins from the wellness aisle.
Treatment for Pulmonary LCH
In adults with lung-predominant disease, smoking cessation is a major part of treatment. In some people, stopping smoking can stabilize the disease and reduce progression risk. Additional treatment may include medications for inflammation, systemic therapy for progressive disease, or targeted therapy in selected cases.
When lung damage is advanced, long-term respiratory monitoring becomes important. In severe and unusual situations, transplant evaluation may enter the conversation.
Supportive and Long-Term Care
Some problems caused by LCH can persist even after the active disease is controlled. For example:
- Diabetes insipidus may require long-term hormone replacement
- Bone damage may need orthopedic support
- Dental or jaw disease may require specialized care
- Lung disease may need pulmonary follow-up
- Neurocognitive or endocrine issues may need long-term monitoring
That is why successful LCH care often involves a team: oncology, endocrinology, pulmonology, dermatology, orthopedics, radiology, and sometimes dentistry or neurology.
Outlook and Prognosis
The prognosis for Langerhans cell histiocytosis varies widely. People with single-system LCH, especially isolated bone disease, often do very well. Those with multisystem disease, particularly when the liver, spleen, or bone marrow are affected, may face a more complicated course.
Relapse is possible, even after a good initial response. Some patients develop long-term effects from the disease itself rather than the active cell growth, including hormone problems, bone changes, hearing issues, or chronic lung disease. Because of that, follow-up is not just a formality. It is part of treatment.
The encouraging news is that outcomes have improved significantly with earlier diagnosis, better imaging, more standardized treatment plans, and newer targeted therapies. In other words, the LCH story is no longer as bleak as it once was, even though it still deserves serious attention.
Real-World Experiences With LCH: What the Journey Often Feels Like
For patients and families, the experience of LCH is often less like a straight road and more like a confusing GPS route that keeps saying, “Recalculating.” The first challenge is usually not treatment. It is getting the right diagnosis.
A parent may notice that a toddler has a scalp rash that just will not clear up, even after creams and shampoo changes. Then there is a soft spot on the skull, or recurrent ear drainage, or unusual thirst that seems out of proportion. An X-ray, biopsy, and referral later, the family is hearing a diagnosis they have never encountered before. That unfamiliarity alone can be overwhelming. Rare diseases have a way of making people feel like they have been handed a manual written in a language they do not speak.
Adults can have a different but equally frustrating path. Someone may develop a chronic cough and shortness of breath and assume it is asthma, bronchitis, or smoking-related irritation. Imaging may reveal lung cysts or nodules, and only after a more detailed evaluation does pulmonary LCH come into view. For others, bone pain or a rash may trigger months of specialist visits before the pieces finally fit together.
Once treatment starts, experiences vary depending on disease severity. A person with a single bone lesion may undergo a procedure and move into follow-up fairly quickly. A child with multisystem disease may need repeated clinic visits, scans, blood work, medication schedules, and long stretches of uncertainty. Families often describe the emotional rhythm as strange: one week feels almost normal, and the next revolves around test results, steroid side effects, or waiting for a scan report to post.
There is also the issue of invisible stress. Even when a patient “looks fine,” LCH can affect sleep, school, work, appetite, mood, and family routines. Steroids can cause irritability or appetite changes. Hormone problems can linger. Parents may worry about relapse every time a new symptom appears. Adult patients may struggle with smoking cessation, fatigue, or anxiety about lung function. This is not just a medical condition on paper; it reshapes daily life in very practical ways.
Many people living with LCH also become accidental experts. They learn how to track symptoms, organize records, interpret scan language, and ask sharper questions at appointments. They find out that “stable disease” can be very good news. They also discover that recovery is not always about one dramatic finish line. Sometimes it is about steady management, fewer symptoms, better energy, and a little more breathing room in everyday life.
One of the most important lessons from real-world LCH experience is that support matters. Patients do better when care is coordinated, when symptoms are taken seriously early, and when families understand that follow-up is part of the process rather than proof that something is going wrong. Rare disease care can feel isolating, but informed care teams and clear communication make a real difference.
In practical terms, the journey often includes many small victories: a child returning to school, a scan showing no new lesions, improved thirst after treatment, a smoker successfully quitting, or a family finally hearing that the disease is under control. None of those moments are flashy. All of them matter.
When to See a Doctor
Because LCH is rare, most everyday rashes or aches are not going to turn out to be this condition. Still, medical evaluation is important when symptoms are unusual, persistent, or unexplained. Seek medical care if there is:
- Ongoing bone pain or swelling
- A persistent rash that does not improve with standard treatment
- Unexplained excessive thirst and urination
- Chronic cough or shortness of breath, especially in smokers
- Repeated ear drainage, jaw problems, or unexplained loose teeth
- Weight loss, fatigue, enlarged lymph nodes, or abdominal swelling
Early evaluation does not guarantee an LCH diagnosis, of course, but it does improve the odds of catching serious conditions before they dig in deeper.
Conclusion
Langerhans cell histiocytosis (LCH) is a rare but important disorder that can affect bones, skin, lungs, the pituitary gland, and other organs. Its symptoms are wide-ranging, which is one reason diagnosis can take time. Researchers now understand that LCH is often driven by mutations in pathways that control cell growth, and that insight has helped expand treatment beyond older one-size-fits-all approaches.
The most effective treatment depends on the patient’s age, the organs involved, and whether the disease is isolated or multisystem. Some people need only local treatment and monitoring. Others need chemotherapy, targeted therapy, hormone management, or long-term specialty follow-up. The good news is that modern care has improved outcomes, especially when the disease is recognized early and managed by experienced teams.
If there is one takeaway worth underlining, circling, and maybe sticking to the refrigerator with a magnet, it is this: LCH may be rare, but persistent, unexplained symptoms deserve a real evaluation. Sometimes the body whispers before it shouts.